2011 AMBER Tutorial with Biotin and Streptavidin
For additional Rizzo Lab tutorials see AMBER Tutorials.
In this tutorial, we will learn how to run a molecular dynamics simulation of a protein-ligand complex. We will then post-process that simulation by calculating structural fluctuations (with RMSD) and free energies of binding (MM-GBSA).
Contents
Biotin Notes
Biotin is also called vitamin H. And it takes part in multiple processes inside the cell. It's a B-complex vitamin (coenzyme) that's involved in gluconeogenesis, citric acid cycle, and various carboxylation reactions.
Streptavidin Notes
Download PDB Here and view it's details Here. Streptavidin has an incredibly strong affinity for biotin; the dissociation constant for the streptavidin-biotin complex is on the order of femtomolar.
....
....
What is AMBER?
Amber - Assisted Model Building with Energy Refinement - is a suite of about 50 programs that can be used to simulate, study and analyze macromolecular systems such as proteins dissolved in water at physiological conditions. Amber10, the current version (Amber11 soon to be released) of Amber, is extremely advanced, powerful and fast. PMEMD, particle mesh Ewald MD (boundary condition treatment / parallelized code) can churn out 314 ps/day of data for the system dihydrofolate reductase (159 residue protein) in TIP3P water (23,558 total atoms). However, because PMEMD lacks the ability to restrain the atoms we need properly, we will be using SANDER to perform most of our simulations.
AMBER Notes
The Amber 10 Manual is the primary resource when trying to learn what variables and keywords mean and what they do. Using Adobe Acrobat to view the file, you can simply search the document for keywords, which saves much time.
Keywords for preparatory programs:
LEaP: creates or modifies systems in Amber. It consists of the functions of prep, link, edit, and parm.
ANTECHAMBER: the main Antechamber suite program that helps prepare input files for nucleic acids and proteins for LEaP.
Keywords for simulating programs:
SANDER: according to the Amber 10 manual, it is 'a basic energy minimizer and molecular dynamics program. This program relaxes the structure by iteratively moving the atoms down the energy gradient until a sufficiently low average gradient is obtained. The molecular dynamics portion generates configurations of the system by integrating Newtonian equations of motion. MD will sample more configurational space than minimization, and will allow the structure to cross over small potential energy barriers. Configurations may be saved at regular intervals during the simulation for later analysis, and basic free energy calculations using thermodynamic integration may be performed. More elaborate conformational searching and modeling MD studies can also be carried out using the SANDER module. This allows a variety of constraints to be added to the basic force field, and has been designed especially for the types of calculations involved in NMR structure refinement'.
PMEMD: verison of SANDER that allows parallel scaling and optimized speed.
There is a mailing list you could sign-up for, as an additional resource.
Unix tips
This is specific to our cluster (seawulf) and desktop (mathlab) environments. See Activating your Seawulf Account for details of sw short cut.
Download Files from SeaWulf to Herbie:
ssh compute.mathlab.sunysb.edu
Login in to Herbie
mkdir sw_dir
make a directory "sw_dir" for which to download files and be organized
cd to sw_dir so when you scp files or directories back to Herbie, it copies them to a specific directory - "sw_dir"
cd sw_dir
scp -r sw:/location_of_files_or_directory/ .
Safely Copy, Recursively, /location_of_files_or_directory/
run.sander.MPI.csh
include these lines before mpirun command to know which nodes mpi is running on
echo "Queue is giving this nodes:" cat \$PBS_NODEFILE echo "MPI is running on:" mpirun -n 8 hostname
Structure Preparation
To begin with, create the directories in seawulf you will work in, using the commands here:
mkdir AMBER_Tutorial cd AMBER_Tutorial cd ~rizzo/AMBER_Tutorial/000.AMBERFILES . mkdir 001.CHIMERA.MOL.PREP mkdir 002.TLEAP mkdir 003.SANDER mkdir 004.ptraj
Copy the commands above to your terminal and hit enter one at a time.
Our next step will be to process a pdb file into receptor, ligand, and complex so that we have will separate files which will eventually be used to setup a molecular dynamics simulation.
Open Chimera, choose File - Fetch by ID, then type in "1df8". Now you will see your protein and ligand in Chimera.
1. It is a dimer, but you need only a monomer.
- Click Select - chain - B, you would see chain B is highlighted.
- Then click Action - Atoms/Bonds - delete.
Now only a receptor, a ligand and several water molecules are left.
2. Now you need to separate the ligand and receptor.
- First, Select - residue - HOH, then delete it.
- File - Save PDB, save this pdb as "1df8.rec.lig.pdb", then Select - residue - BTN, delete it. Save PDB as "1df8.rec.noh.pdb."
- Second, open the 1df8.rec.lig.pdb, select the receptor and delete it (Tips: you can invert your selection.)
- Then Tools - structure editing - Add H, press OK.
- Then Tools - structure editing -
- Add Charge, press OK (use any charge model at this point).
- Then Select AM1-BCC charge model and hit OK.
This will assign to your ligand the AM1-BCC charge model.
- File - Save mol2, save it as "1df8.lig.chimera.mol2".
- Open up 1df8.lig.chimera.mol2 with your favorite text editor (i.e vim). Manually change atom names to be unique. The simplest way is to append a number after each atom label. Save the new file as 1df8.lig.mol2. This has to be done because tleap only uses the first 3 characters as the name and each atom in a given residue must be unique.
1df8.lig.chimera.mol2:
1 C11 32.5640 18.1390 14.0710 C.2 1 BTN1 0.9079
2 O11 33.4260 17.4630 13.4730 O.co2 1 BTN1 -0.8596
3 O12 32.5320 19.3920 13.9660 O.co2 1 BTN1 -0.8472
4 C10 31.5990 17.4500 14.9620 C.3 1 BTN1 -0.1966
5 C9 31.2260 16.0460 14.5600 C.3 1 BTN1 -0.0459
6 C8 30.5160 16.0360 13.2000 C.3 1 BTN1 -0.0920
7 C7 30.0160 14.6260 12.8220 C.3 1 BTN1 -0.0683
8 C2 29.2080 14.5510 11.5450 C.3 1 BTN1 -0.0028
9 S1 27.5110 15.2280 11.7030 S.3 1 BTN1 -0.2811
10 C6 27.1670 14.6500 10.0230 C.3 1 BTN1 -0.0520
11 C5 27.7360 13.2480 9.9740 C.3 1 BTN1 0.0662
12 N1 26.8850 12.1810 10.4970 N.pl3 1 BTN1 -0.4731
1df8.lig.mol2
1 C1 32.5640 18.1390 14.0710 C.2 1 BTN1 0.9079
2 O2 33.4260 17.4630 13.4730 O.co2 1 BTN1 -0.8596
3 O3 32.5320 19.3920 13.9660 O.co2 1 BTN1 -0.8472
4 C4 31.5990 17.4500 14.9620 C.3 1 BTN1 -0.1966
5 C5 31.2260 16.0460 14.5600 C.3 1 BTN1 -0.0459
6 C6 30.5160 16.0360 13.2000 C.3 1 BTN1 -0.0920
7 C7 30.0160 14.6260 12.8220 C.3 1 BTN1 -0.0683
8 C8 29.2080 14.5510 11.5450 C.3 1 BTN1 -0.0028
9 S9 27.5110 15.2280 11.7030 S.3 1 BTN1 -0.2811
10 C10 27.1670 14.6500 10.0230 C.3 1 BTN1 -0.0520
11 C11 27.7360 13.2480 9.9740 C.3 1 BTN1 0.0662
12 N12 26.8850 12.1810 10.4970 N.pl3 1 BTN1 -0.4731
3. At this point we will copy over the contents of AMBER_Tutorial to the Seawulf cluster to run antechamber, tleap, and sander programs. Note that to copy files you must be on a portal to seawulf (i.e. silver, herbie or ringo). If user is on a MATHLAB machine then files are accessible from herbie (also called compute). Therefore login to herbie and copy the files over using:
scp -r AMBER_Tutorial sw:
- This copies the entire folder AMBER_Tutorial (and subfolders) from herbie to your top directory on seawulf
- Note that these commands only apply to the AMS536 class at Stony Brook (or Rizzo lab rotation students) and is specific for our computer setups. Users outside the University, on other systems, will need to use use slightly different commands
TLEAP
Class please add in a Tleap section here.
Class I've gotten a section started please improve this section. Thanks.
describe our protocol and give example input files.
Note: The following sections should be done on while you are on the Seawulf cluster.
Go to the 002.TLEAP directory
cd ~/AMBER_Tutorial/002.TLEAP
Next we will use vi to make three input files which will be used by TLEAP to create parameter/topology files and initial coordinate files for (1) the ligand, (2) the receptor, and (3) the complex. Lets make file #1. Use vi and make a file called "tleap.lig.in". Cut and paste the following into the file and save it.
set default PBradii mbondi2 source leaprc.ff99SB loadoff ions.lib loadamberparams ions.frcmod source leaprc.gaff loadamberparams gaff.dat.rizzo loadamberparams 1df8.lig.ante.frcmod loadamberprep 1df8.lig.ante.prep lig = loadpdb 1df8.lig.ante.pdb saveamberparm lig 1df8.lig.gas.leap.prm7 1df8.lig.gas.leap.rst7 solvateBox lig TIP3PBOX 10.0 saveamberparm lig 1df8.lig.wat.leap.prm7 1df8.lig.wat.leap.rst7 charge lig quit
Repeat this process for file#2 called "tleap.rec.in"
set default PBradii mbondi2 source leaprc.ff99SB loadoff ions.lib loadamberparams ions.frcmod REC = loadpdb ../001.CHIMERA.MOL.PREP/1df8.rec.noh.pdb saveamberparm REC 1df8.rec.gas.leap.prm7 1df8.rec.gas.leap.rst7 solvateBox REC TIP3PBOX 10.0 saveamberparm REC 1df8.rec.wat.leap.prm7 1df8.rec.wat.leap.rst7 charge REC quit
Do the same for a file#3 called "tleap.com.in"
set default PBradii mbondi2
source leaprc.ff99SB
loadoff ions.lib
loadamberparams ions.frcmod
source leaprc.gaff
loadamberparams gaff.dat.rizzo
REC = loadpdb ../001.CHIMERA.MOL.PREP/1df8.rec.noh.pdb
loadamberparams 1df8.lig.ante.frcmod
loadamberprep 1df8.lig.ante.prep
LIG = loadpdb 1df8.lig.ante.pdb
COM = combine {REC LIG}
saveamberparm COM 1df8.com.gas.leap.prm7 1df8.com.gas.leap.rst7
solvateBox COM TIP3PBOX 10.0
saveamberparm COM 1df8.com.wat.leap.prm7 1df8.com.wat.leap.rst7
charge COM
quit
Now we will make a csh script (also called a shell script) which is used to setup all necessary files used for molecular dynamics calculations. Note that many commands in so-called shell scripts can be performed manually on the command line but we use scripts to makes the process easier. Note that the csh script will use the three files we mmade above. make a file called "run.TLEAP.csh" and cut and paste the following into the file and save it.
#! /bin/tcsh set workdir = "~/AMBER_Tutorial/002.TLEAP" cd $workdir rm 1df8.* ANTECHAMBER* *.out ATOMTYPE.INF leap.log NEWPDB.PDB PREP.INF cp ../000.AMBERFILES/* . antechamber -i ../001.CHIMERA.MOL.PREP/1df8.lig.mol2 -fi mol2 -o 1df8.lig.ante.pdb -fo pdb antechamber -i ../001.CHIMERA.MOL.PREP/1df8.lig.mol2 -fi mol2 -o 1df8.lig.ante.prep -fo prepi parmchk -i 1df8.lig.ante.prep -f prepi -o 1df8.lig.ante.frcmod tleap -s -f tleap.lig.in > tleap.lig.out tleap -s -f tleap.rec.in > tleap.rec.out tleap -s -f tleap.com.in > tleap.com.out ambpdb -p 1df8.com.gas.leap.prm7 -tit "pdb" <1df8.com.gas.leap.rst7 > 1df8.com.gas.leap.pdb exit
Make sure that the three programs you want to run are accessible by your environment by using the "which" command:
which antechamber which tleap
If the programs are not found please let your TA know.
To run this script you will issue the following command:
csh run.TLEAP.csh
It is essential that you fully understand the various commands performed when you execute the above shell script. For example, making directories, copying files, running executables, etc.
The primary purpose of the script is to assign force field parameters to each of the three species. For example, antechamber is run first to create a pdb and then a prepi file, which contains the internal coordinates of the ligand. Then parmchk is run to create a frcmod file, which contains additional parameters not in GAFF and needed by tleap. Look at your input and output files for ALL tleap calculations with your favorite text editor (eg. vim) to be sure you understand what is being done.
Note that for the receptor processing four residues (22, 27, 40, and 57) have two possible conformers, when tleap builds your parameter/topology and coordinate files only the first occurrence of each residue is retained. See section below and leap output.
To insure that TLEAP ran correctly and generated all necessary files issue the following:
ls
You should have all of the following files:
1df8.com.gas.leap.prm7 1df8.rec.gas.leap.rst7 ions.lib 1df8.com.gas.leap.rst7 1df8.rec.wat.leap.prm7 leap.log 1df8.com.wat.leap.prm7 1df8.rec.wat.leap.rst7 NEWPDB.PDB 1df8.com.wat.leap.rst7 ANTECHAMBER_AC.AC parm.e16.dat 1df8.lig.ante.frcmod ANTECHAMBER_AC.AC0 PREP.INF 1df8.lig.ante.pdb ANTECHAMBER_BOND_TYPE.AC run.TLEAP.csh 1df8.lig.ante.prep ANTECHAMBER_BOND_TYPE.AC0 tleap.com.in 1df8.lig.gas.leap.prm7 ANTECHAMBER_PREP.AC tleap.com.out 1df8.lig.gas.leap.rst7 ANTECHAMBER_PREP.AC0 tleap.lig.in 1df8.lig.wat.leap.prm7 ATOMTYPE.INF tleap.lig.out 1df8.lig.wat.leap.rst7 gaff.dat.rizzo tleap.rec.in 1df8.rec.gas.leap.prm7 ions.frcmod tleap.rec.out
Visualization:
On Herbie, copy the prmtop and rst files from seawulf to Herbie:
cd ~/AMBERTutorial/002.TLEAP/ scp sw:~/AMBERTutorial/002.TLEAP/\*.rst7 . scp sw:~/AMBERTutorial/002.TLEAP/\*.prm7 .
Note that we had to escaping the "*" with "\*". This is because the "*" has a function which stands for wild card on the current machine (herbie). The "\*" is the escaping the function and is just the passing the character the the other machine (seawulf).
run VMD on your desktop.
Main window File->New Molecule load prm7 solvated complex then load rst7 solvated complex
LETS INSERT SOME FIGURES HERE.
Minimization and equilibration
In order to adjust our structures to the force field and remove any model building artifacts, we first perform a several-step equilibration protocol. Several iterations of minimization and molecular dynamics will be preformed with decreasing restraints.
The first step: Relaxing the experimental or silico structure
Write the following to file 01mi.in:
01mi.in: equilibration &cntrl imin = 1, maxcyc = 1000, ntmin = 2, ntx = 1, ntc = 1, ntf = 1, ntb = 1, ntp = 0, ntwx = 1000, ntwe = 0, ntpr = 1000, scee = 1.2, cut = 8.0, ntr = 1, restraintmask = ':1-119 & !@H=', restraint_wt=5.0, /
lets try and run a minimization
1. request a processor (on a node) to run your jobs on from the queue (i.e. log onto a node interactively).
qsub -I -l nodes=1:ppn=1
2. next you will run a serial version of sander on the processor by performing the following command on the command line.
sander -O -i 01mi.in -o 01mi.out -p ../002.TLEAP/1df8.com.wat.leap.prm7 \ -c ../002.TLEAP/1df8.com.wat.leap.rst7 -ref ../002.TLEAP/1df8.com.wat.leap.rst7 \ -x 01mi.trj -inf 01mi.info -r 01mi.rst
3. exit the node.
4. Now lets submit this same job remotely by doing the following:
Write the tcsh file test1.qsub.csh
#! /bin/tcsh
#PBS -l nodes=1:ppn=1
#PBS -l walltime=720:00:00
#PBS -o zzz.qsub.out
#PBS -e zzz.qsub.err
#PBS -V
set workdir = "~/AMBER_Tutorial/003.SANDER"
cd ${workdir}
cat $PBS_NODEFILE | sort | uniq
sander -O -i 01mi.in -o 01mi.out -p ../002.TLEAP/1df8.com.wat.leap.prm7 \
-c ../002.TLEAP/1df8.com.wat.leap.rst7 -ref ../002.TLEAP/1df8.com.wat.leap.rst7 \
-x 01mi.trj -inf 01mi.info -r 01mi.rst
exit
Make this file executable by doing the following:
chmod +x test1.qsub.csh
Now submit the job to the queue
qsub test1.qsub.csh
5. Now lets run perellel sander:
Write the tcsh file test2.qsub.csh
#! /bin/tcsh
#PBS -l nodes=2:ppn=2
#PBS -l walltime=720:00:00
#PBS -o zzz.qsub.out
#PBS -e zzz.qsub.err
#PBS -V
set workdir = "~/AMBER_Tutorial/003.SANDER"
cd ${workdir}
cat $PBS_NODEFILE | sort | uniq
mpirun -n 4 sander.MPI -O -i 01mi.in -o 01mi.out -p ../002.TLEAP/1df8.com.wat.leap.prm7 \
-c ../002.TLEAP/1df8.com.wat.leap.rst7 -ref ../002.TLEAP/1df8.com.wat.leap.rst7 \
-x 01mi.trj -inf 01mi.info -r 01mi.rst7
exit
Make this file executable by doing the following:
chmod +x test2.qsub.csh
Now submit the job to the queue
qsub test2.qsub.csh
Write the following to file 10md.in:
10md.in: production (500000 = 1ns) &cntrl imin = 0, ntx = 5, irest = 1, nstlim = 500000, temp0 = 298.15, tempi = 298.15, ig = 71287, ntc = 2, ntf = 1, ntt = 1, dt = 0.002, ntb = 2, ntp = 1, tautp = 1.0, taup = 1.0, ntwx = 500, ntwe = 0, ntwr = 500, ntpr = 500, scee = 1.2, cut = 8.0, iwrap = 1, ntr = 1, nscm = 100, restraintmask = ':1-118@CA,C,N', restraint_wt = 0.1, /
The following are our exacted equilibration and production protocols.
equilibration:
01mi.in: maxcyc = 1000, restraintmask = ':1-119 & !@H=', restraint_wt = 5.0,
02md.in: ntx = 1, irest = 0, nstlim = 50000, restraintmask = ':1-119 & !@H=',
restraint_wt = 5.0, dt = 0.001,
03mi.in: maxcyc = 1000, restraintmask = ':1-119 & !@H=', restraint_wt = 2.0,
04mi.in: maxcyc = 1000, restraintmask = ':1-119 & !@H=', restraint_wt = 0.1,
05mi.in: maxcyc = 1000, restraintmask = ':1-119 & !@H=', restraint_wt = 0.05,
06md.in: ntx = 1, irest = 0, nstlim = 50000, restraintmask = ':1-119 & !@H=',
restraint_wt = 1.0, dt = 0.001,
ntwx = 1000, ntwr = 1000, ntpr = 1000,
07md.in: nstlim = 50000, restraintmask = ':1-119 & !@H=', restraint_wt = 0.5, dt = 0.001,
ntwx = 1000, ntwr = 1000, ntpr = 1000,
08md.in: nstlim = 50000, restraintmask = ':1-118@CA,C,N', restraint_wt = 0.1, dt = 0.001,
ntwx = 1000, ntwr = 1000, ntpr = 1000,
09md.in: nstlim = 50000, restraintmask = ':1-118@CA,C,N', restraint_wt = 0.1, dt = 0.001,
ntwx = 1000, ntwr = 1000, ntpr = 1000,
production:
10md.in: nstlim = 500000, restraintmask = ':1-118@CA,C,N', restraint_wt = 0.1, dt = 0.002, 11md.in: nstlim = 500000, restraintmask = ':1-118@CA,C,N', restraint_wt = 0.1, dt = 0.002,
Note that the other parameters not specified should be the same as those in the above two files for minimization and molecular dynamics, respectively. copy and modify 01mi.in for the minimization inputs and 10md.in for the Molecular Dynamics inputs.
Now, lets run the equilibration and production.
Write the tcsh file equil.produc.qsub.csh
#! /bin/tcsh
#PBS -l nodes=4:ppn=2
#PBS -l walltime=720:00:00
#PBS -o zzz.qsub.out
#PBS -e zzz.qsub.err
#PBS -V
set workdir = "~/AMBER_Tutorial/003.SANDER"
cd ${workdir}
cat $PBS_NODEFILE | sort | uniq
mpirun -n 8 sander.MPI -O -i 01mi.in -o 01mi.out -p ../002.TLEAP/1df8.com.wat.leap.prm7 \
-c ../002.TLEAP/1df8.com.wat.leap.rst7 -ref ../002.TLEAP/1df8.com.wat.leap.rst7 \
-x 01mi.trj -inf 01mi.info -r 01mi.rst7
mpirun -n 8 sander.MPI -O -i 02md.in -o 02md.out -p ../002.TLEAP/1df8.com.wat.leap.prm7 \
-c 01mi.rst7 -ref 01mi.rst7 -x 02md.trj -inf 02md.info -r 02md.rst7
mpirun -n 8 sander.MPI -O -i 03mi.in -o 03mi.out -p ../002.TLEAP/1df8.com.wat.leap.prm7 \
-c 02md.rst7 -ref 02md.rst7 -x 03mi.trj -inf 03mi.info -r 03mi.rst7
mpirun -n 8 sander.MPI -O -i 04mi.in -o 04mi.out -p ../002.TLEAP/1df8.com.wat.leap.prm7 \
-c 03mi.rst7 -ref 03mi.rst7 -x 04mi.trj -inf 04mi.info -r 04mi.rst7
mpirun -n 8 sander.MPI -O -i 05mi.in -o 05mi.out -p ../002.TLEAP/1df8.com.wat.leap.prm7 \
-c 04mi.rst7 -ref 04mi.rst7 -x 05mi.trj -inf 05mi.info -r 05mi.rst7
mpirun -n 8 sander.MPI -O -i 06md.in -o 06md.out -p ../002.TLEAP/1df8.com.wat.leap.prm7 \
-c 05mi.rst7 -ref 05mi.rst7 -x 06md.trj -inf 06md.info -r 06md.rst7
mpirun -n 8 sander.MPI -O -i 07md.in -o 07md.out -p ../002.TLEAP/1df8.com.wat.leap.prm7 \
-c 06md.rst7 -ref 05mi.rst7 -x 07md.trj -inf 07md.info -r 07md.rst7
mpirun -n 8 sander.MPI -O -i 08md.in -o 08md.out -p ../002.TLEAP/1df8.com.wat.leap.prm7 \
-c 07md.rst7 -ref 05mi.rst7 -x 08md.trj -inf 08md.info -r 08md.rst7
mpirun -n 8 sander.MPI -O -i 09md.in -o 09md.out -p ../002.TLEAP/1df8.com.wat.leap.prm7 \
-c 08md.rst7 -ref 05mi.rst7 -x 09md.trj -inf 09md.info -r 09md.rst7
mpirun -n 8 sander.MPI -O -i 10md.in -o 10md.out -p ../002.TLEAP/1df8.com.wat.leap.prm7 \
-c 09md.rst7 -ref 05mi.rst7 -x 10md.trj -inf 10md.info -r 10md.rst7
mpirun -n 8 sander.MPI -O -i 11md.in -o 11md.out -p ../002.TLEAP/1df8.com.wat.leap.prm7 \
-c 10md.rst7 -ref 05mi.rst7 -x 11md.trj -inf 11md.info -r 11md.rst7
exit
Make this file executable by doing the following:
chmod +x equil.produc.qsub.csh
Now submit the job to the queue
qsub equil.produc.qsub.csh
To see that you have successfully submitted your job and monitor its progress uses the following:
qstat
or
qstat -u username
sander input parameters
- &cntrl -> Tells SANDER that what follows are control variables.
- imin=1 -> Perform Minimization
- maxcyc=1000 -> Perform 1000 Minimization Steps
- ntmin=2 -> Steepest Descent Method of Minimization
- ntx=1 -> Initial Coordinates Lack Velocity - it's a restart file (See VMD)
- ntc=1 -> "SHAKE" Posititional Restraints OFF (Default)
- ntf=1 -> Calculate All types of Forces (bonds, angles, dihedrals, non-bonded)
- ntb=1 -> Constant Volume Boundary Periodicity
- ntp=0 -> No Pressure Regulation
- ntwx=1000 -> Print Coordinates Frequency
- ntwe=0 -> Print Energy to "mden" Frequency
- ntpr=1000 -> Print Readable Energy Information to "mdout" and "mdinfo"
- scee' -> 1-4 Coulombic Forces are Divided (Default=1.2)
- cut=8.0 -> Coulombic Force Cutoff distance in Angstroms
- restraintmask = ':1-119 & !@H=', -> restraint the residues matching the mask':1-119 & !@H='. Here, we're restraining residues 1 through 119 and everything that isn't hydrogen. Essentially, onlt Hydrogen atoms move free of restraint.
- restraint_wt=5.0 is the Force Constant assigned to the restrained atoms. Each atom "sits" in a potential-energy well characterized by a "5.0" kcal/mol wall.
- / is used to the machine to stop the job when it's done.
Sander Flags
A Production simulation is the final simulation to be performed. Once the structure is built (See tLeap section), minimized and equilibrated (See Minimization and equilibration section) and only essential restraints retained, production dynamics produce the data used to answer the scientific question at hand. The previous steps were just preparatory.
The important instructions are the 18th and 19th lines - mpirun.
mpirun - Instructs SeaWulf (SW) to use an mpi regime.
-n - Denotes number of processors (Here 8)
sander.MPI - Is the actual MPI code to be run. This is the heart of the whole course. The text after this and on this line will be instructions for sander.
-O Overwrite previous output files with the new ones (to be produced by this job). This is useful in that if you have to run this script more than once, then something must have gone wrong. Thus, this will simply overwrite the files that are erroneous anyways. Don't use this if you really know what your doing and have a reasonable reason for doing so.
-i Points sander to the input file (10md.in then 11md.in).
-o Instructs sander to write the output file (10md.out then 11md.out), which contains the energies written by sander during dynamics. The various variables within the input file will determine the nature of the output: How frequently are the energies printed? How detailed is the information? What types of energies are being printed?
-p Points sander to the topology (aka parameter file). See Amber10 manual if you don't know what this is.
-c Points sander to the coordinates (corresponding to the topology file from above) for which you want the production dynamics to begin from. In this case, we're starting with the final (snapshot - single frame) frame from the previous (equilibration) run, 9md.rst. The .rst at the end of a file means "restart". "restart" files are single frames. They can contain subtle information in addition to simply the number of atoms in the file (first line) and the x, y and z coordinates for each atom (which is why you need the topology file to give those x,y,z's meaning - what atom has what coordinates) so check the Amber10 manual if you're curious.
-ref Specifies for sander which file you would like the restraints (if not using restraints, then -ref is not used) to be centered on. The reference is a snapshot-set of coordinates which you have carefully chosen to represent the "ideal" structure you would like the dynamics (sander) to use as a "reference"; using the restrain_wt option in the input file, you're telling sander how much like the reference structure you want your dynamical system to be. The restraint_wt option acts like a spring, where harmonically the reference coordinates and coordinates of the dynamical system are "psuedo-connected." Thus, it is imperative that the reference structure be realistic (say the coordinates of the crystal structure) and be the same system (i.e. same number of atoms).
-x Instructs sander to print the trajectory (position of each atom along with its velocity) every ntwx steps. This is the "Big" file. When your simulation is complete, zip the trajectories:
gzip 10md.trj gzip 11md.trj
The trajectories are, in my opinion, the most important component of a MD experiment. So, read the Amber10 Manual. You could notice that ptraj uses the trajectory files to perform the bulk of the data analysis like RMSD, H-bonding evolution, radius of gyration, pi-stacking, etc., etc.
-info Gives the results of the interactions between the supercomputer and sander: How did the calculation proceed? Did everything work properly? Did the simulation run to completion? This flag can be useful in debugging failed jobs....hk...
-r Instructs sander to write a restart file. The frequency this is done is specified is ntwr in the 10md.in / 11md.in files. Usually ntwr=500. A restart file will be written at the end of the simulation - the final snapshot of the simulation will be the restart file if and when the simulation has run successfully to completion. During the simulation, however, this file is continuously re-written as a fail-safe - say the supercomputer crashes during your 10ns simulation, which has been running for 3 weeks. Well, the restart file is printed every ntwr steps so you could, as the name of the flag implies, simply restart the simulation (with minor modification to the input file and above runsander.csh script) from the last snapshot before the crash.
Check results
When your simulations have finished, you ought to check the stability and realism of results. Use the script E_asis to analyze the the mdout files. This ought to also be used to check the validity and stability of your production runs.
Download E_asis onto your local machine (the one you're using right now). Once saved onto local machine, transfer it to you working directory on Herbie, Seawulf, etc. Follow usual protocols to do this. This script will extract the energy, temperature, pressure and volume (and averages thereof) from the mdout file. To execute, do the following (it may be a good idea to make a separate directory just for this analysis, as many files are created):
chmod +x E_asis
./E_asis Filename.out
Filenames, as per this tutorial ought to be 01min, 02md, 03md, etc. To analyze the whole equilibration experiment (i.e. 01min to 09md), the following may work. Please check the results to be sure it worked properly. There are various ways to coordinate the analysis of these output files..
./E_asis *.out
ptraj - Analyzing Your Data
We will use ptraj, a program that is distributed with AMBER Tools, for our analysis. It is simple to do the same analysis with VMD, and you may wish to do so and include it in the tutorial.
You will want to create a directory called 004.PTRAJ
mkdir 004.PTRAJ
Then go the directory you just created
cd 004.PTRAJ
In that directory, you will need to create the following input file called ptraj.1.in containing the following lines:
trajin ../003.SANDER/10md.trj 1 1000 1 trajin ../003.SANDER/11md.trj 1 1000 1 trajout 1df8.trj.strip nobox strip :WAT
This will concatenate your two 1ns trajectories together, strip off the waters, and output it as a new file called 1df8.trj.strip.
To execute, in the command line write:
ptraj ../002.TLEAP/1df8.com.wat.leap.prm7 ptraj.1.in > ptraj.1.log
Note: Because we are piping the ptraj output in the .log file, always check this file afterwards to ensure everything when smoothly. If something isn.t found, check your paths, and if something wasn.t executed correctly, check your commands.
Now create a new file called ptraj.2.in containing the following lines:
trajin 1df8.trj.strip 1 2000 1 trajout 1df8.com.trj.stripfit reference ../002.TLEAP/1df8.com.gas.leap.rst7 rms reference out 1df8.rmsd.CA.txt :1-118@CA
To execute, in the command line write:
ptraj ../002.TLEAP/1df8.com.gas.leap.prm7 ptraj.2.in > ptraj.2.log
This will align your trajectory using the positions of the receptor.s alpha carbons, and output a file containing the rmsd of the alpha carbons, and new trajectory fit aligned using those atoms. . If you open the text file, you will see two columns of data, the frame in the first column and the rmsd in the second. You can use any graphing software to create a plot of the rmsd of the alpha carbons. This should be very stable, because we restrained these in our simulation (see 10md.in and 11md.in).
For our final rmsd analysis, create a file called ptraj.3.in containing:
trajin 1df8.com.trj.stripfit 1 2000 1 reference ../002.TLEAP/1df8.com.gas.leap.rst7 rms reference out 1df8.lig.rmsd.txt :119@C*,N*,O*,S* nofit
To execute:
ptraj ../002.TLEAP/1df8.com.gas.leap.prm7 ptraj.3.in > ptraj.3.log
This will create a txt file containing the rmsd of the ligand to the original crystal structure (as represented by our reference, 1df8.com.gas.leap.rst7). Again, graph it, and look for deviations in the rmsd. Visualize the trajectory using vmd (loading in the stripfit trajectory as an AMBER coordinate file using the 1df8.com.gas.leap.prm7 file would be the most expeditious route) and identify what the major changes are. You may wish to do further analysis about distances between certain atoms or the presence of hydrogen bonds using VMD or ptraj (see link below).
Finally, we will want to perform MMGBSA analysis on our trajectories. To do this, we will need a trajectory with our complex and no waters (which we created above called 1df8.com.trj.stripfit), and will want to create two more trajectories containing just the receptor and just the ligand. To do so, we.ll need two more ptraj inputs:
ptraj.4.in containing:
trajin 1df8.com.trj.stripfit 1 2000 1 trajout 1df8.rec.trj.stripfit strip :119
and ptraj.5.in containing:
trajin 1df8.com.trj.stripfit 1 2000 1 trajout 1df8.lig.trj.stripfit strip :1-118
To execute, type in the following commands:
ptraj ../002.TLEAP/1df8.com.gas.leap.prm7 ptraj.4.in > ptraj.4.log ptraj ../002.TLEAP/1df8.com.gas.leap.prm7 ptraj.5.in > ptraj.5.log
Also, you can write a csh file to go through the procedure above.
Make a file analy.1.csh in your AMBER_tutorial directory as follows:
#! /bin/tcsh mkdir 004.PTRAJ cd ./004.PTRAJ cat << EOF > ptraj.1.in trajin ../003.SANDER/10md.trj 1 1000 1 trajin ../003.SANDER/11md.trj 1 1000 1 trajout 1df8.trj.strip nobox strip :WAT EOF ptraj ../002.TLEAP/1df8.com.wat.leap.prm7 ptraj.1.in >ptraj.1.log cat << EOF > ptraj.2.in trajin 1df8.trj.strip trajout 1df8.com.trj.stripfit reference ../002.TLEAP/1df8.com.gas.leap.rst7 rms reference out 1df8.rmsd.CA.txt :1-118@CA EOF ptraj ../002.TLEAP/1df8.com.gas.leap.prm7 ptraj.2.in >ptraj.2.log cat << EOF > ptraj.3.in trajin 1df8.com.trj.stripfit reference ../002.TLEAP/1df8.com.gas.leap.rst7 rms reference out 1df8.lig.rmsd.txt :119@C*,N*,O*,S* nofit EOF ptraj ../002.TLEAP/1df8.com.gas.leap.prm7 ptraj.3.in >ptraj.3.log cat << EOF > ptraj.4.in trajin 1df8.com.trj.stripfit trajout 1df8.rec.trj.stripfit strip :119 EOF cat <<EOF > ptraj.5.in trajin 1df8.com.trj.stripfit trajout 1df8.lig.trj.stripfit strip :1-118 EOF ptraj ../002.TLEAP/1df8.com.gas.leap.prm7 ptraj.4.in >ptraj.4.log ptraj ../002.TLEAP/1df8.com.gas.leap.prm7 ptraj.5.in >ptraj.5.log cd ..
Then use "csh" command to execute the file analy.1.in in your AMBER_tutorial directory.
Making xmgrace plots
Write out a tcsh script to run xmgrace
foreach distance (28OH_119O14 12OG_119O14) cat << EOF > $distance.grace.in READ NXY "$distance.out" s0 legend "$distance" title "Hbond Distance" xaxis label "time (ps)" yaxis label "Hbond distance (angstroms)" PRINT TO "$distance.eps" DEVICE "EPS" OP "level2" PRINT EOF xmgrace -batch $distance.grace.in -nosafe -hardcopy end
This will generate two eps plots for the hydrogen bond distances.
Hydrogen bond distance
Before using ptraj to measure the H-bond distance, it's better to know which atoms we want to put in the ptraj script. VMD can automatically draws these H-bonds. First, select the ligand and residues around the ligand. Second, change the Drawing Method to HBonds.
After we know the atom name, we use distance command to measure the distance between two atoms. Here is the sample script
ptraj ../002.TLEAP/1df8.com.gas.leap.prm7 << EOF trajin ../004.PTRAJ/1df8.com.trj.stripfit distance 34N_119O2 :34@N :119@O2 out 34N_119O2.out distance 73OG_119O3 :73@OG :119@O3 out 73OG_119O3.out distance 12OG_119O14 :12@OG :119@O14 out 12OG_119O14.out distance 28OH_119O14 :28@OH :119@O14 out 28OH_119O14.out EOF
Plot the result. Here is the sample. It will be a good way to compare the H-bond distance with the binding free energy from MM-GBSA.
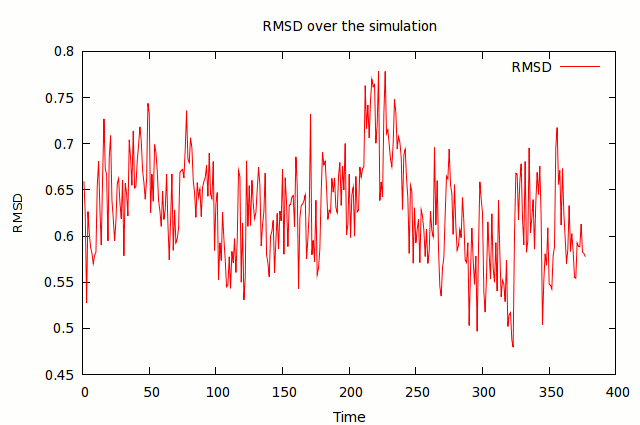
MMGBSA
Move up one directory and create the following directory:
mkdir 005.MMGBSA
In this new directory, you will want to create this final file called “gb.rescore.in” containing:
Single point GB energy calc &cntrl ntf = 1, ntb = 0, ntc = 2, idecomp= 0, igb = 5, saltcon= 0.00, gbsa = 2, surften= 1.0, offset = 0.09, extdiel= 78.5, cut = 99999.0, nsnb = 99999, scnb = 2.0, scee = 1.2, imin = 5, maxcyc = 1, ncyc = 0, /
You will then want to create a csh script that contains the following lines of command (called run.sander.rescore.csh)
#! /bin/tcsh
#PBS -l nodes=1:ppn=2
#PBS -l walltime=24:00:00
#PBS -o zzz.qsub.out
#PBS -e zzz.qsub.err
#PBS -V
set workdir = “${HOME}/AMBER_Tutorial/005.MMGBSA”
cd ${workdir}
sander -O -i gb.rescore.in \
-o gb.rescore.out.com \
-p ../002.TLEAP/1df8.com.gas.leap.prm7 \
-c ../002.TLEAP/1df8.com.gas.leap.rst7 \
-y ../004.PTRAJ/1df8.com.trj.stripfit \
-r restrt.com \
-ref ../002.TLEAP/1df8.com.gas.leap.rst7 \
-x mdcrd.com \
-inf mdinfo.com
sander -O -i gb.rescore.in \
-o gb.rescore.out.lig \
-p ../002.TLEAP/1df8.lig.gas.leap.prm7 \
-c ../002.TLEAP/1df8.lig.gas.leap.rst7 \
-y ../004.PTRAJ/1df8.lig.trj.stripfit \
-r restrt.lig \
-ref ../002.TLEAP/1df8.lig.gas.leap.rst7 \
-x mdcrd.lig \
-inf mdinfo.lig
sander -O -i gb.rescore.in \
-o gb.rescore.out.rec \
-p ../002.TLEAP/1df8.rec.gas.leap.prm7 \
-c ../002.TLEAP/1df8.rec.gas.leap.rst7 \
-y ../004.PTRAJ/1df8.rec.trj.stripfit \
-r restrt.rec \
-ref ../002.TLEAP/1df8.rec.gas.leap.rst7 \
-x mdcrd.rec \
-inf mdinfo.rec
exit
Then you should qsub this script:
chmod +x run.sander.rescore.csh qsub run.sander.rescore.csh
After this job has run, you will obtain your output files: gb.rescore.out.com, gb.rescore.out.lig, and gb.rescore.out.rec. In these files, the single point energy calculation results will be written out for each individual frame. It can be found in the results section and should look like this:
FINAL RESULTS
NSTEP ENERGY RMS GMAX NAME NUMBER
1 7.9662E+03 1.9504E+01 1.3272E+02 C 3403
BOND = 836.7674 ANGLE = 2343.1208 DIHED = 2917.9356
VDWAALS = -2299.0570 EEL = -19265.6215 EGB = -3539.4050
1-4 VDW = 1071.1234 1-4 EEL = 11631.2887 RESTRAINT = 0.0000
ESURF = 14270.0746
minimization completed, ENE= 0.79662270E+04 RMS= 0.195036E+02
minimizing coord set # 2
You will need to obtain the necessary results and perform simple calculations on them. First, to easily obtain the results (without doing a LOT of copying or deleting) is to use the grep command. Note, if you are familiar with python or perl, it would be simplest to write a little program to read in these files and obtain these values.
For the following set of commands, you’ll need to do this for com, lig, and rec. In the command line, type
grep VDWAALS gb.rescore.out.com > vdw.com.txt grep ESURF gb.rescore.out.com > surf.com.txt
You can take these text files, import them into excel, and do the rest of your modifications there.
Remember that to obtain the Gvdw term, you need to take the SASA (which is ESURF) and input into equation 1:
ΔGnonpolar = SASA*0.00542 + 0.92 [1]
Also, the mmgbsa of a given system can be determined by equation 2:
ΔGmmgbsa = ΔGvdw + ΔGcoul + ΔGpolar + ΔGnonpolar [2]
From the output file:
VDWAALS = ΔGvdw
EELS = ΔGcoul
EGB = ΔGpolar
You can then easily calculate the ΔΔGbind by using equation 3:
ΔΔGbind = ΔGmmgbsa,complex – (ΔGmmgbsa,lig + ΔGmmgbsa,rec) [3]
You will want to careful when doing your analysis that the results from frame 1 for the receptor and ligand are subtracted from the results from frame 1 for your complex. By doing this in excel, you should have 2000 frames for each, and the values should cleanly line up. Finally, you will want to plot your ΔΔGbind and examine if you see corresponding changes in the ligand position and the ΔΔGbind. Also, you should determine the mean and standard deviation for your ΔΔGbind.
Please remember, you are required to post your analysis and the process by which you obtained your results on the wiki. If you use different programs or a different approach or do any further analysis, please share with your peers. When posting, be clear, and concise but thorough.
Change "run.sander.rescore.csh" into an executable
chmod +x run.sander.rescore.csh
Submit the run.sander.rescore.csh to the queue
qsub run.sander.rescore.csh
Monitor your job
qstat -u YourUserName
Data extraction and calculation
When your restoring calculation finishes, you have the following three output files: "gb.rescore.out.com", "gb.rescore.out.lig", and "gb.rescore.out.rec".
Use the following script, entitled get.mmgbsa.bash, to extract data and calculate MMGBSA energy for each snap shot.
#! /bin/bash
# by Haoquan
echo com lig rec > namelist
LIST=`cat namelist`
for i in $LIST ; do
grep VDWAALS gb.rescore.out.$i | awk '{print $3}' > $i.vdw
grep VDWAALS gb.rescore.out.$i | awk '{print $9}' > $i.polar
grep VDWAALS gb.rescore.out.$i | awk '{print $6}' > $i.coul
grep ESURF gb.rescore.out.$i | awk '{print $3 * 0.00542 + 0.92}' > $i.surf
paste -d " " $i.vdw $i.polar $i.surf $i.coul | awk '{print $1 + $2 + $3 + $4}' > data.$i
rm $i.*
done
paste -d " " data.com data.lig data.rec | awk '{print $1 - $2 - $3}' > data.all
for ((j=1; j<=`wc -l data.all | awk '{print $1}'`; j+=1)) do
echo $j , >> time
done
paste -d " " time data.all > MMGBSA_vs_time
rm namelist time data.*
Run this script:
bash get.mmgbsa.bash
Now you have the final data sheet: MMGBSA_vs_time. Copy them to excel or Origin and separate two columns by comma.